-1024x684.jpg)
-1024x683.jpg)
食品合成着色剂,也称为食品合成染料,被广泛应用到食品的加工过程中。鉴于合成着色剂的不安全性,国家加强了对这类合成着色剂的监管,制订了食品合成着色剂的测定标准(GB5009.35-2016)。
逗点生物依据国标的征求意见稿,建立了固相萃取-高效液相色谱法测定食品中8种合成着色剂含量的方法,样品经乙醇氨水提取后,经Copure® PWAX混合型弱阴离子交换柱净化,采用高效液相色谱仪结合紫外检测器测定。
经验证,8种合成着色剂在0.10 μg/mL到10 μg/mL范围内线性良好,R^2>0.995,加标回收率在80%-110 %范围内,符合测试需求。

一、样本前处理
1.1样品提取
准确称取样品2 g(精确至0.001 g),置于50 mL具塞离心管中,加入25 mL乙醇氨水溶液(硬糖需要先加入2 mL水超声使其溶解),涡旋1 min,50℃超声提取20 min,8000 转/分钟离心5 min。
取上清液置于50 mL离心管中,残渣中再加入15 mL乙醇氨水溶液重复提取1次,离心后合并上清液,用乙醇氨水溶液定容至50 mL,即得提取液。
准确吸取提取液10 mL,50℃下氮气浓缩至2 mL左右,分2~3次共加入10 mL 5%甲醇水溶液溶解,作为待净化液。
注:乙醇氨水溶液配制,量取无水乙醇700 mL,加入4 mL氨水,用水稀释并定容至1 L,混匀。

1.2试样的净化浓缩
先依次用6 mL甲醇和6 mL水活化Copure® WAX固相萃取柱(150 mg/6 mL),保持柱体湿润,将待净化液注入柱中,控制流出液流速在1滴/秒,弃去流出液。再依次用6 mL 2%甲酸水和6 mL甲醇淋洗,弃去淋洗液,抽干小柱。
最后用6 mL 2%氨化甲醇洗脱,分两次加入,收集洗脱液,于50 ℃氮吹至近干。准确加入2 mL pH=9的乙酸铵溶液溶解,过0.45 μm PTFE滤膜,弃去2~5滴初滤液,取续滤液作为待测液。
1.3空白实验
1.3.1过程空白:
取一支洁净的50 mL具塞离心管,不称取试样,按照上述1.1和1.2进行提取、净化,浓缩,取续滤液上机测试。
1.3.2空白本底实验
称取样品2 g(精确至0.001 g)按照上述1.1和1.2进行提取、净化,浓缩,取续滤液上机测试。
二、 标准系列工作液


精密移取着色剂混标中间液(1 μg/mL)0.10 mL、0.20 mL于2个1 mL色谱瓶中,再吸取混合标准中间液(10 μg/mL)0.05 mL、0.10 mL、0.20 mL、0.50mL于3个1 mL色谱瓶中,分别用水定容至1 mL,混匀,得标准系列工作液。
浓度分别为0.10 μg/mL、0.20 μg/mL、0.50 μg/mL、1.00 μg/mL、2.00 μg/mL、5.00 μg/mL和10.0 μg/mL。
三、仪器条件
仪器:ThermoFisher U3000液相色谱仪
色谱柱:CommaSil® C18
(4.6 mm×250 mm,5 μm)
流动相:A:20 mmol/L乙酸铵溶液 B:甲醇
洗脱方式:梯度洗脱,见表1
流速:1 Ml/min
柱温:30 ℃
进样量:10 Μl
检测器:紫外检测器

四、实验结果
4.1加标回收
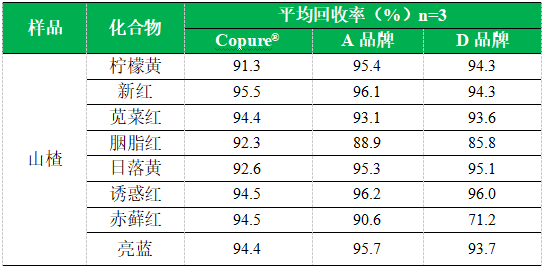
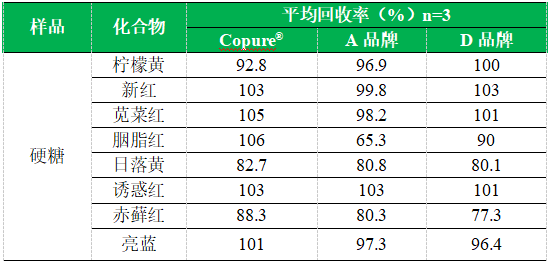
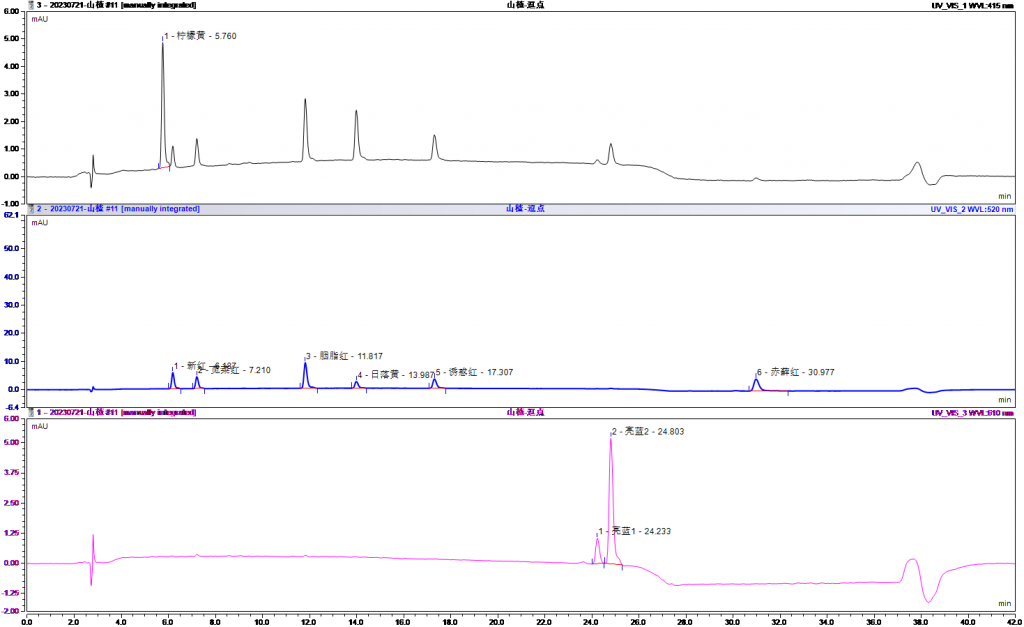
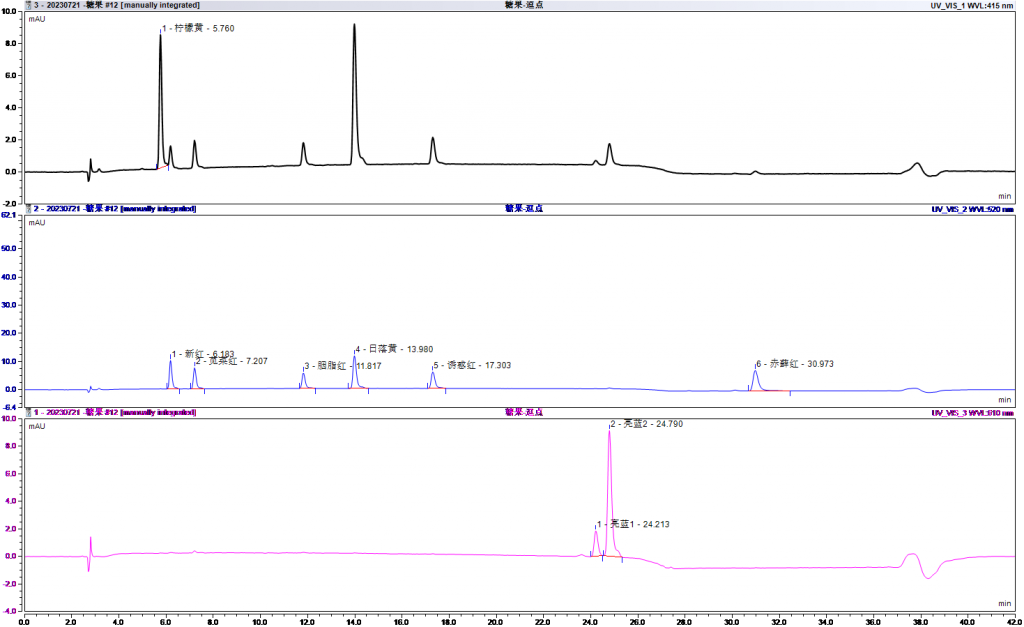
五、注意事项
1)在使用乙醇氨水溶液提取时,需要提取两次以上,提取一次可能会导致提取不充分,导致回收率偏低。
2)过柱前确认样液的pH值<6,以便于合成着色剂目标化合物和填料PWAX更好的结合。
3)上机之前过滤所用的针式过滤器不能使用尼龙针式过滤器,会吸附部分合成着色剂,导致回收率偏低,建议使用PTFE亲水针式过滤器。
4)在检测赤藓红时,需要注意在使用离心管氮吹时会有少量的赤藓红会附着在离心管上,可以采用涡旋和超声的方法进行溶解,使用玻璃的氮吹管更易溶解;或者在溶解的时候加入一定量的甲醇便于赤藓红的溶解。
六、订购信息
